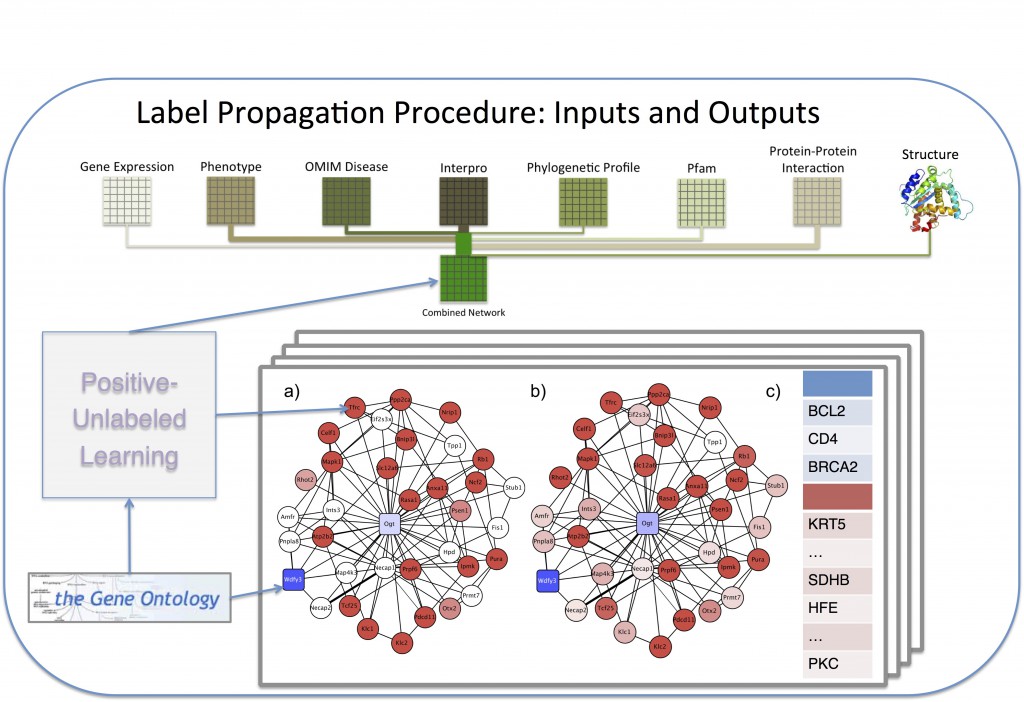
The rate of new protein discovery has, in recent years, outpaced our ability to annotate and characterize new proteins and proteomes. In order to combat this functional annotation deficit, many groups have successfully turned to computational techniques, attempting to predict the function of proteins in order to guide experimental verification. The most prolific methods come from machine learning, and have been applied to the task of predicting the biological function of proteins based on a variety of available data. The majority of these methods require negative examples: proteins that are known not to perform a function, in order to achieve meaningful predictions, but negative examples are often not available. Thus we combine a machine-learning approach, Gaussian Random Field Label Propagation, with a semi-supervised paradigm where only the positive examples are labeled, known as Positive-Unlabled learning.
Our novel techniques have been applied in the scope of the Critical Assessment of Functional Annotation (CAFA) challenge, annotating more than 100,000 sequences across 27 species. These annotations incorporate data from gene expression, protein-protien interaction, Interpro databases, phylogenetic profiles, existing GO annotations, and protein structure. For tertiary structure, we employ a new approach including deNovo folding of sequences using Rosetta, as well as structural similarity between existing known structures, combined at the domain level in order to maximize the functional information gain. Lastly, we incorporate homology edges between species networks, calculated using Inparanoid scores. This allows us to propagate annotations across species, even if there are no existing training annotations in the annotations for a particular organism.
The results of our work will appear in the Automated-Function-Prediction meeting at the 2014 ISMB conference in July.